 Bioinformatics research in the department of computer science focuses on the study of proteins using computational techniques. This involves the development of novel techniques and algorithms for structural analysis of proteins, ther interactions and interfaces. Developing solutions to such problems in Bioinformatics require a complete understanding of their biological foundations as well as computational science. Such research involves the use of advanced methods in machine learning such as kernel based and deep learning, data mining, big data analytics, high performance computing and algorithms. Bioinformatics research at DCIS works in close collaboration with leading biological research centers both within and outside Pakistan and covers a broad domain of applied topics. Such topics have a direct impact on biological discovery through bioinformatics in understanding how viruses work within the human body and how to design new proteins from scratch with the use of computing technology.
Bioinformatics research in the department of computer science focuses on the study of proteins using computational techniques. This involves the development of novel techniques and algorithms for structural analysis of proteins, ther interactions and interfaces. Developing solutions to such problems in Bioinformatics require a complete understanding of their biological foundations as well as computational science. Such research involves the use of advanced methods in machine learning such as kernel based and deep learning, data mining, big data analytics, high performance computing and algorithms. Bioinformatics research at DCIS works in close collaboration with leading biological research centers both within and outside Pakistan and covers a broad domain of applied topics. Such topics have a direct impact on biological discovery through bioinformatics in understanding how viruses work within the human body and how to design new proteins from scratch with the use of computing technology.
If you plan to join our research labs to work on Bioinformatics, you will be involved in the development of cutting edge tools in this area for use by biologists. You will get a flavor of the complete spectrum of advanced Computer Science topics in an environment conducive to exploration and active learning. Armed with this experience, you can go on to become a data scientist for any national or international facility that works in Bioinformatics. Our MS and PhD programs in this area focus on personal development and enable every student to be at the forefront of biological science and computing technology.
People
Research
Prediction of Protein Interfaces
We work on the development of machine learning based methods for predicting protein interfaces and binding sites. The basic approach is to use data from known protein complexes to train a machine learning model to predict how two previously unseen proteins bind to each other. In this domain, we have developed two different approaches which are briefly explained below.
PAIRpred
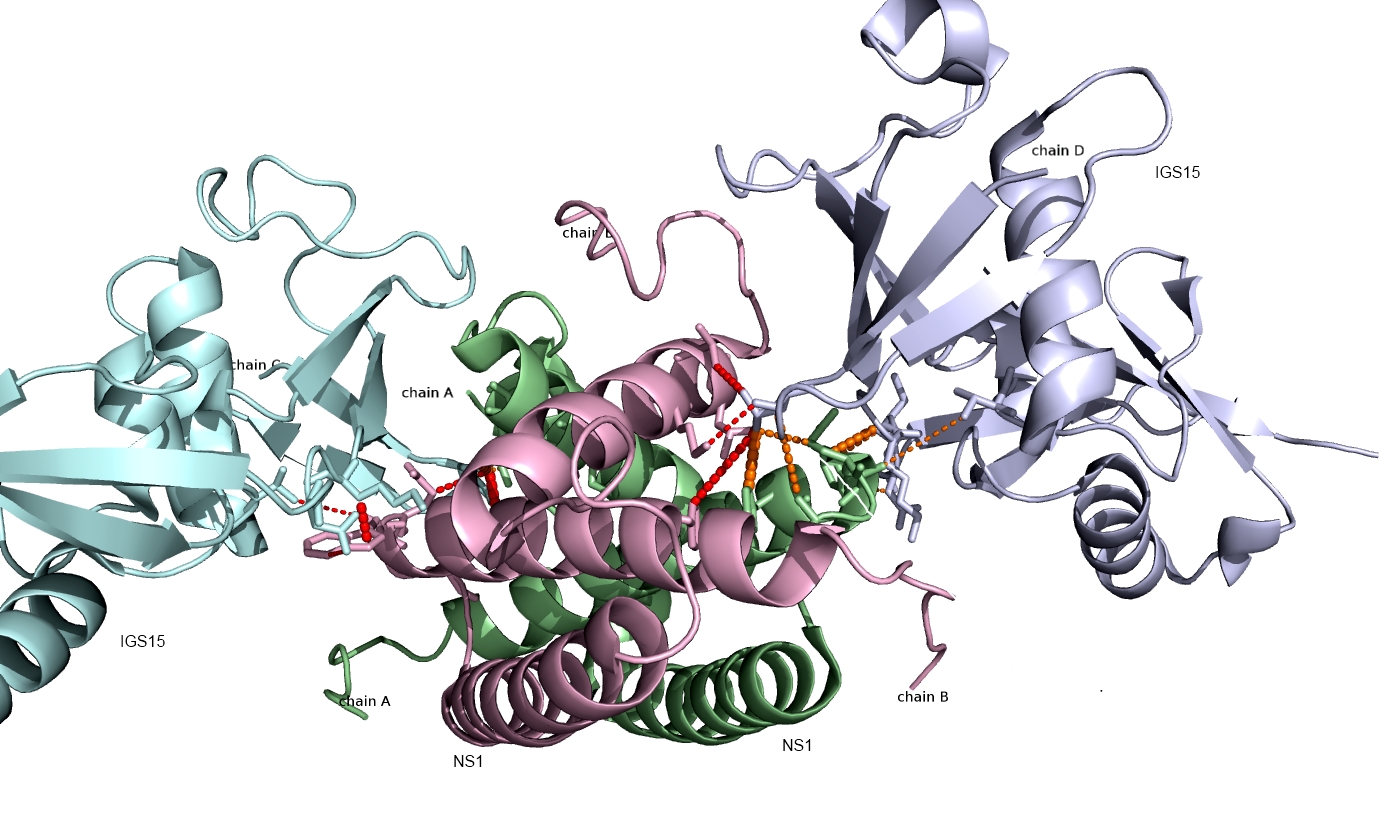
PAIRPred (Partner Aware Interacting Residue PREDictor) is a partner specific protein-protein interaction site predictor that can make accurate predictions of whether a pair of residues from two different proteins interact or not. It differs from most existing interaction site predictors in that it considers the information about the interaction partner of a protein in making its predictions whereas most other methods produce partner-independent predictions. It employs a Support Vector Machine (SVM) to generate interaction propensity scores for a pair of residues from sequence information alone or in conjunction with structure based features. PAIRPred offers state of the art prediction accuracy. More details about how PAIRPred works and its performance evaluation are available here. We are also focusing on predicting interfaces between different proteins from Flaviviridae (the virus family includes Dengue, Hepatitis, West Nile, etc.). This work has been published in Proteins.
Shown below are the interactions (orange and red lines) between human ISG15 protein and the NS1 protein from Influenza A virus predicted using PAIRpred. These predictions agree well with experimental findings and have been used to infer why Influenza A infects only humans and non-human primates.
MI-1: Multiple instance learning of Calmodulin binding proteins
Calmodulin is a very important protein found, virtually conserved, across all higher organisms. It is involved in a large number of very critical biological functions ranging from neuronal spiking to breathing. It interacts with a large number of other proteins. Together with Dr. Asa Ben-Hur, I was involved in the development of a sequence based predictor of binding sites on proteins that bind Calmodulin using machine learning. The interesting part of this work was that the annotated binding sites of Calmodulin binding proteins were imprecise and spanned an area larger than the true binding site. Consequently, we developed a novel multiple instance learning method for learning from such noisy and imprecise data. Our algorithm is called MI-1 and is currently the state of the art predictor of binding interfaces in Calmodulin binding proteins. This work has been published in Bioinformatics. The web server for this application can be accessed here.
RAMClust: Clustering of metabolomic data
Metabolomics is the study of unique chemical fingerprints that are left behind by different cellular processes. RAMClust is a technique that uses clustering to group the features resulting from Liquid Chromatography and tandem Mass Spectrometry analysis of metabolomic samples such as urine, serum, etc. This work has been published in Analytical Chemistry and is being used at the Metabolomics and Proteomics Facility at Colorado State University. We have also filed a provisional patent application for this algorithm through CSU ventures.