Biomedical Informatics Lab¶

Table of Contents
Aims and Objectives¶
Welcome to the Biomedical Informatics lab at Pakistan Institute of Engineering and Applied Sciences, Islamabad, Pakistan. This lab focuses on the development of computing solutions to problems in Biology and Medicine. In particular, we focus on applications of machine learning, data analytics and mining in Bioinformatics, medical image and signal analysis in automated diagnosis systems.
You can read about our current and past research on this page. If you would like to know more, please contact Dr. Fayyaz ul Amir Afsar Minhas.
Bioinformatics Research¶
Ever wondered how we found out that humans share more than 90% of their DNA with chimpanzees? Or how is the DNA of a cell determined in the first place? Have you ever thought about the tree of life and tracing human origins? Are you interested in learning how computers are being used to make drugs and personalized medicine for fighting cancers and viruses? How can we design artifical life?

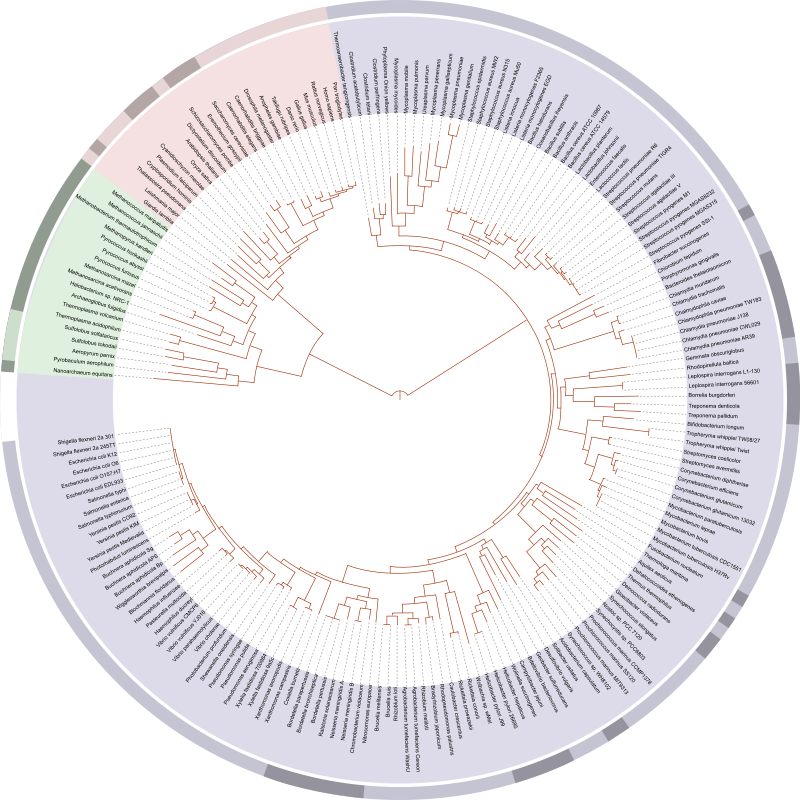

If you find you are a mathematician or a computer scientist who feels intrigued by the above or similar questions, you will appreciate Bioinformatics.
Bioinformatics is an interdisciplinary scientific field that develops mathematical formulations, algorithms and software for producing, storing, retrieving, organizing and analyzing biological data. Let’s see what this means.
Firstly let’s talk about biological data and what it means. Biology has progressed significantly to the point that experts now say that the 21st century belongs to Biology. This has been made possible through tremendous advances in extraction of data from biological systems through technologies such as sequencing, electron microscopy, X-rays, NMR and a lot others. As an example, consider the fact that the number of organisms with sequenced genomes, i.e., organisms whose DNA is now known through sequencing, is growing exponentially. Most biological data is large, noisy and hard or even impossible to interpret for humans without assistance from computers. However, this data is extremely useful. For example, knowing the DNA sequence of an organism allows one to find out, among other things, its evolutionary history, its genes, its disease susceptibility and its genetic behavior towards its environment.
The major problem in Biology is to move from raw data to useful information. This is where computer science comes into play. Solving biological problems through computer science is called Bioinformatics or computational biology. As mentioned earlier, these problems include the storage and retrieval of biological data (because its huge!), its organization and, most importantly, its analysis (because we want to make sense of it!). Research in Bioinformatics has produced algorithms and software which allow the conversion of raw data from biological systems into useful information. For example, sequencing technologies such as next generation sequencing generate a very large number of very short regions (about 500-1000 base pairs) from the very long genome string (about 3.2 Billion base pairs long for human). These reads then need to be mapped and aligned to produce the whole genome. You can think of this as a huge jig-saw puzzle. We can use short read mapping and alignment algorithms to solve this problem. Once the genome is known, we can use phylogenetic tools to construct phylogenetic trees like the tree of life that tell us about the evolutionary origins of an organism and its relationships with other life. We can also use sequencing information and differential expression algorithms to find out what makes a certain variety of crop resistant to drought. Techniques in computational proteomics and structural Bioinformatics can allow us to design new proteins with novel functions to solve global problems such as pollution, energy shortages and diseases. And the best part of it all: Bioinformatics can help in finding out how life works!
Reasons for a computer scientist to be engaged in Bioinformatics research
This is definitely a good time to be a computer scientist and to work in Bioinformatics. Here are some of the reasons why a computer scientist should consider to (or why we do) work in this area:
- Biology easily has 500 years of exciting problems to work on: This quote by Donald Knuth says it all. The analysis of biological data will keep producing more challenging and interesting puzzles that will need computer scientists.
- Most of Bioinformatics is younger than me: Bioinformatics is a relatively young field. As a consequence, there is plenty of energy and lots of challenging unsolved problems or problems needing better solutions.
- Rapidly evolving field: Bioinformatics is an extremely rapidly developing field and it needs more creative solutions from computer scientists.
- Its cross-disciplinary: Bioinformatics is a unique blend of mathematics, statistics, biology and computer science and presents a very fruitful and unique learning experience for researchers.
- Raises interesting computational problems: Due to the nature of biological data, the computational problems encountered in Bioinformatics push the limits of computing and computing technology. The nature and scale of computing solutions required in Bioinformatics allows one to learn the depth and breadth of computer science ranging from algorithms and parallel computing to databases, artificial intelligence and machine learning.
- Its full of very interesting machine learning/data mining problems: Since we know only a little about biological systems and it is really difficult to develop theories about different biological problems, machine learning from biological data finds its home in Bioinformatics. Machine learning is a subfield of computer science and artificial intelligence whose objective is to make computers learn from data. Machine learning systems can generate predictions about biological data and, with help from its half-sibling named data mining, identify important patterns, like say what makes a protein cause a certain disease. Bioinformatics often requires novel and customized machine learning and data mining algorithms for the solution to biological problems.
- Global impact: Due to its importance in Biology and medicine, research in Bioinformatics can have tremendous global impact on health, agriculture, energy and the environment.
- Funding and career opportunities: There is a lot of international funding available for research in Bioinformatics and a large number of career opportunities present themselves to researchers in Bioinformatics.
- Bioinformatics in Pakistan: Bioinformatics is a nascent field in Pakistan and there is plenty of room for it to grow. It is ripe time to start working in Bioinformatics to solve problems tuned to our domestic industries such as agriculture as well as in the fight against diseases such as Dengue. See this page for a very introductory level demonstration on how Bioinformatics can be applied to studying how Dengue works.
Below is a listing of some of the projects we are working on.
Prediction of protein Interfaces and interactions¶
You can think of the biological cell as a chemical factory. In this factory, the genome acts as a pseudo-template for the generation of proteins. These proteins are the workhorse of the cellular factory and are, with a tilt of the hat to DNA and RNA, the most important macromolecules in the cell. Almost all cellular processes directly involve proteins. This can be appreciated by considering the fact that 50% of the dry weight of the human body is protein and 90% of the dry weight of a red blood cell is a single protein called Hemoglobin which is responsible for the transport of Oxygen to cells. Keeping true to our chemical factory analogy, these protein workers work together and interact with each other to perform different cellular functions in the cell. These interactions between proteins are made possible through different physiochemical phenomenon such as hydrogen and covalent bonds, van der Waals forces, the hydrophobic effect etc.
When two protein molecules bind to each other to form a complex, they do so at specific interfaces or binding sites. The study of protein interfaces and binding sites is a very important domain of research in Bioinformatics. Information about the interfaces between proteins can be used not only in understanding protein function but can also be directly employed in drug design and protein engineering. However, the experimental determination of protein interfaces is cumbersome, expensive and not possible in some cases with today’s technology. As a consequence, the computational prediction of protein interfaces from sequence and structure has emerged as a very active research area.
We work on the development of machine learning based methods for predicting protein interactions and interfaces interfaces and binding sites. The basic approach is to use data from known protein complexes to train a machine learning model to predict whether and how two previously unseen proteins bind to each other. Below, we describe the methods developed in this lab together with my collaborators for predicting protein interactions and interfaces.
Host Pathogen Protein Interactions¶
We are currently engaged in research on predicting interactions between host and pathogen proteins. Such interactions form the basis of pathogen borne infectious diseases.
To read more about our work on host pathogen protein interactions, the interested reader is referred to this webpage.
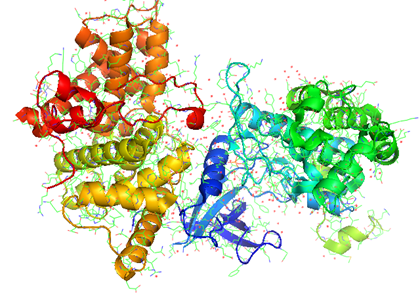
PAIRpred¶
PAIRPred (Partner Aware Interacting Residue PREDictor) is a partner specific protein-protein interaction site predictor that can make accurate predictions of whether a pair of residues from two different proteins interact or not. It differs from most existing interaction site predictors in that it considers the information about the interaction partner of a protein in making its predictions whereas most other methods produce partner-independent predictions. It employs a Support Vector Machine (SVM) to generate interaction propensity scores for a pair of residues from sequence information alone or in conjunction with structure based features. PAIRPred offers state of the art prediction accuracy. More details about how PAIRPred works and its performance evaluation are available here. We are also focusing on predicting interfaces between different proteins from Flaviviridae (the virus family includes Dengue, Hepatitis, West Nile, etc.). This work has been published in Proteins.
Shown below are the interactions (orange and red lines) between human ISG15 protein and the NS1 protein from Influenza A virus predicted using PAIRpred. These predictions agree well with experimental findings and have been used to infer why Influenza A infects only humans and non-human primates.
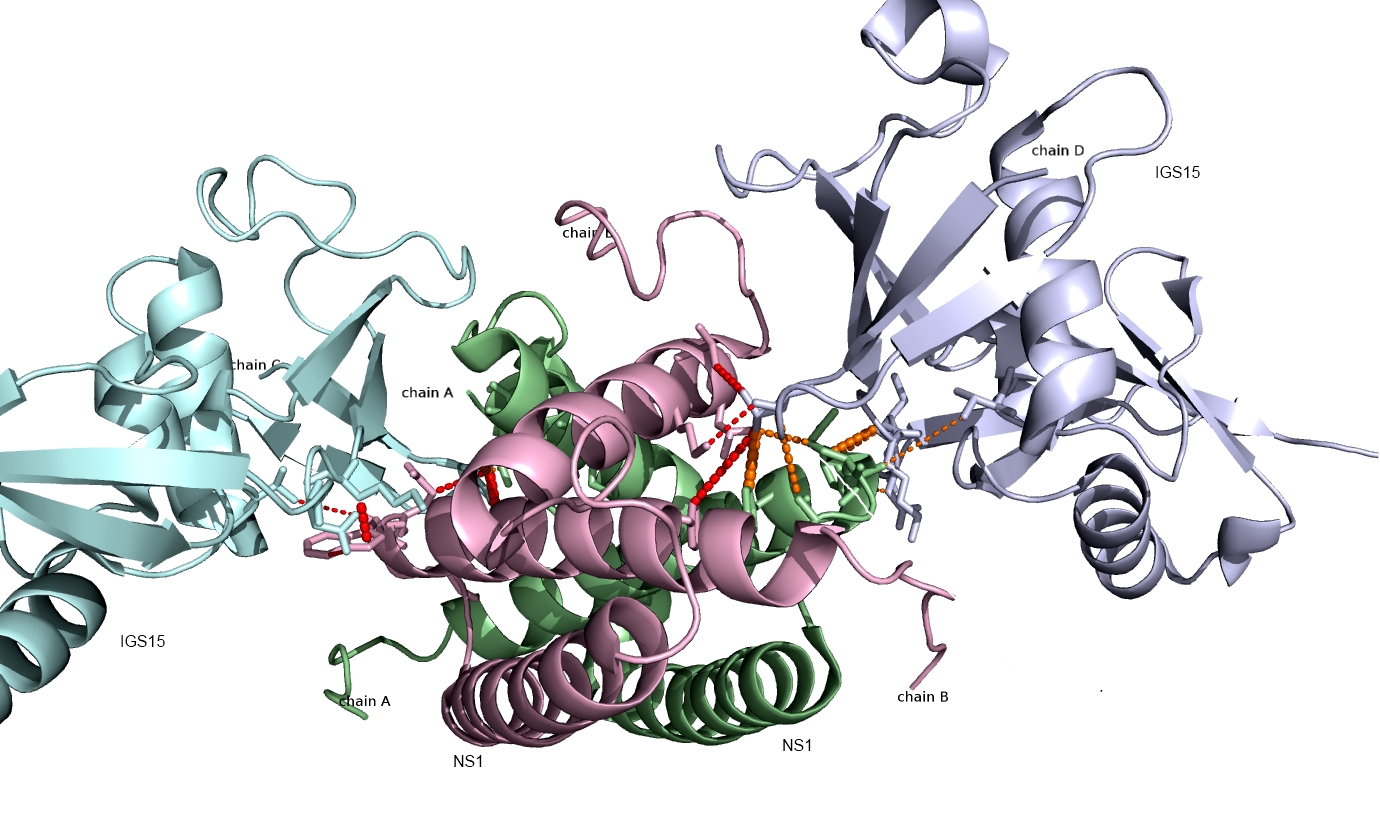
Currently, we are working on developing a webserver for PAIRpred and improving its prediction accuracy for complexes involving large binding associated conformational changes.
MI-1: Multiple instance learning of Calmodulin binding proteins¶
Calmodulin is a very important protein found, virtually conserved, across all higher organisms. It is involved in a large number of very critical biological functions ranging from neuronal spiking to breathing. It interacts with a large number of other proteins. Together with Dr. Asa Ben-Hur, Dr. Minhas was involved in the development of a sequence based predictor of binding sites on proteins that bind Calmodulin using machine learning. The interesting part of this work was that the annotated binding sites of Calmodulin binding proteins were imprecise and spanned an area larger than the true binding site. Consequently, we developed a novel multiple instance learning method for learning from such noisy and imprecise data. Our algorithm is called MI-1 and is currently the state of the art predictor of binding interfaces in Calmodulin binding proteins. This work has been published in Bioinformatics. The web server for this application can be accessed here.
Shown below is an example prediction generated by MI-1. It shows the interaction of Bordetella pertussis adenylyl cyclase toxin (PDB entry: 1yrt) with calmodulin. The binding of adenylate cyclase with CaM is one of the two mechanisms which allows the whooping cough bacteria to colonize the respiratory tract. The colors on adenylyl cyclase (from blue to red) indicate the sequence-only predictions generated by MI-1. The maximum propensity of interaction occurs at W242 (shown in stick form, encircled in green) which is known to be an interaction site of adenylyl cyclase with Calmodulin. This protein was not part of the training set of MI-1.

RAMClust: Clustering of metabolomic data¶
Metabolomics is the study of unique chemical fingerprints that are left behind by different cellular processes. RAMClust is a technique that uses clustering to group the features resulting from Liquid Chromatography and tandem Mass Spectrometry analysis of metabolomic samples such as urine, serum, etc. This work has been published in Analytical Chemistry and is being used at the Metabolomics and Proteomics Facility at Colorado State University. We have also filed a provisional patent application for this algorithm through CSU ventures.
Protein Design and Molecular Dynamics¶
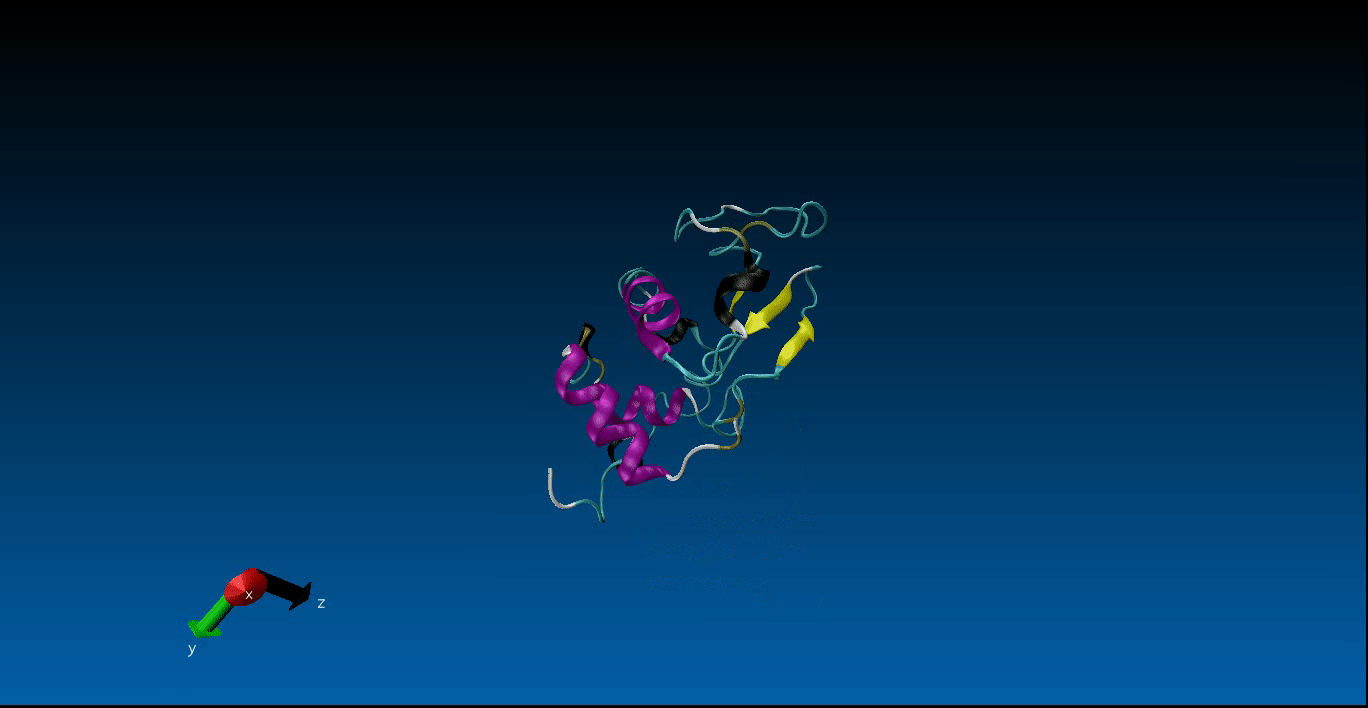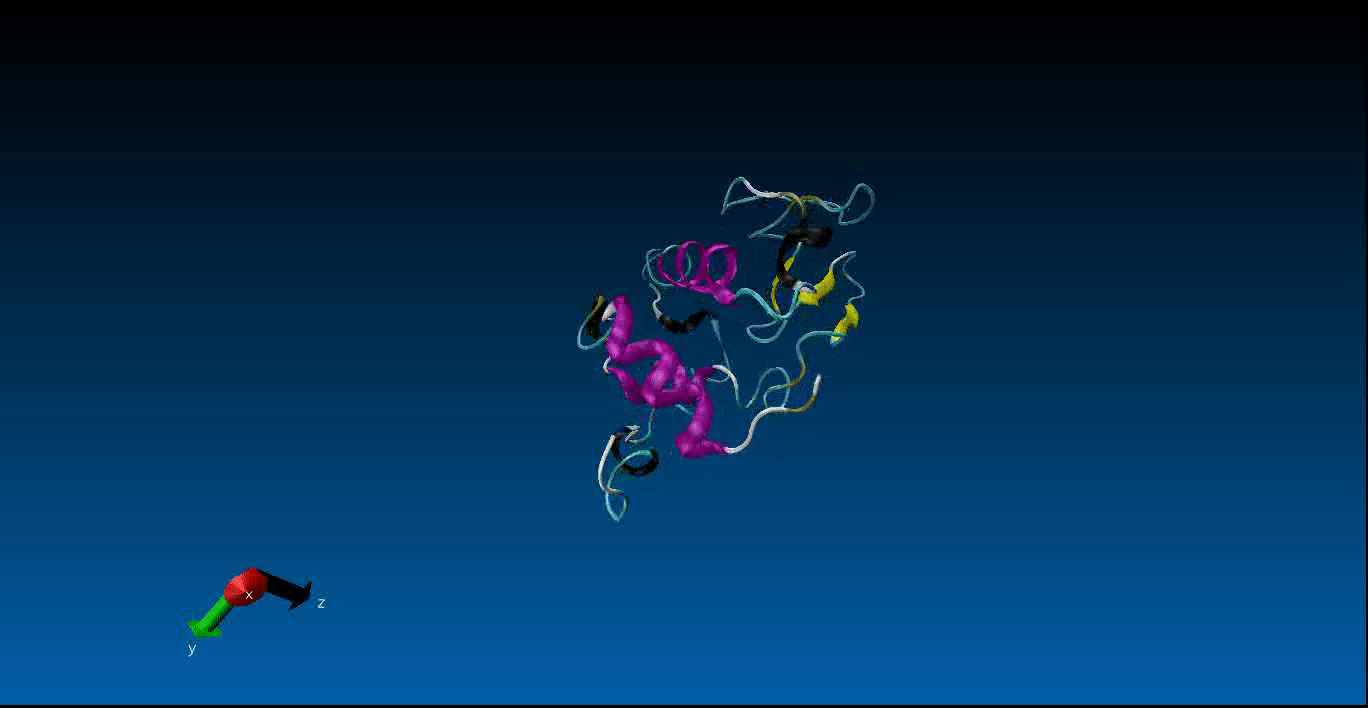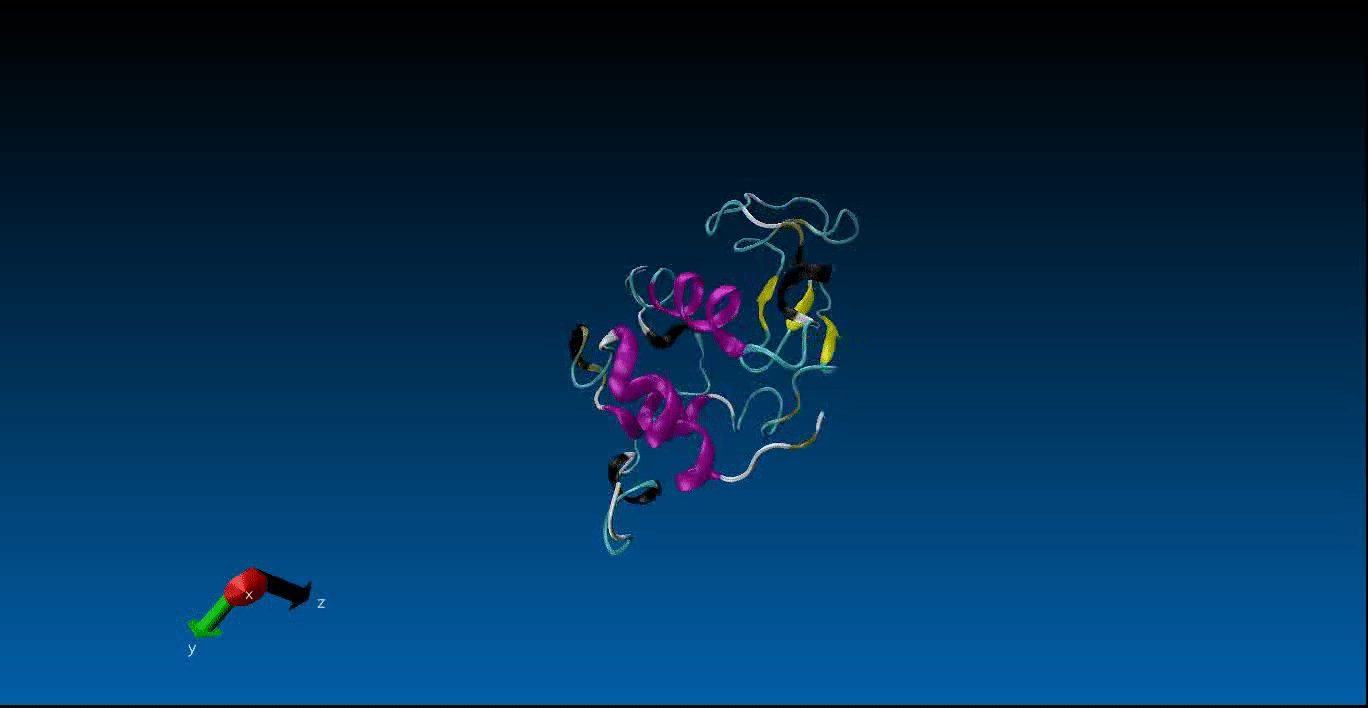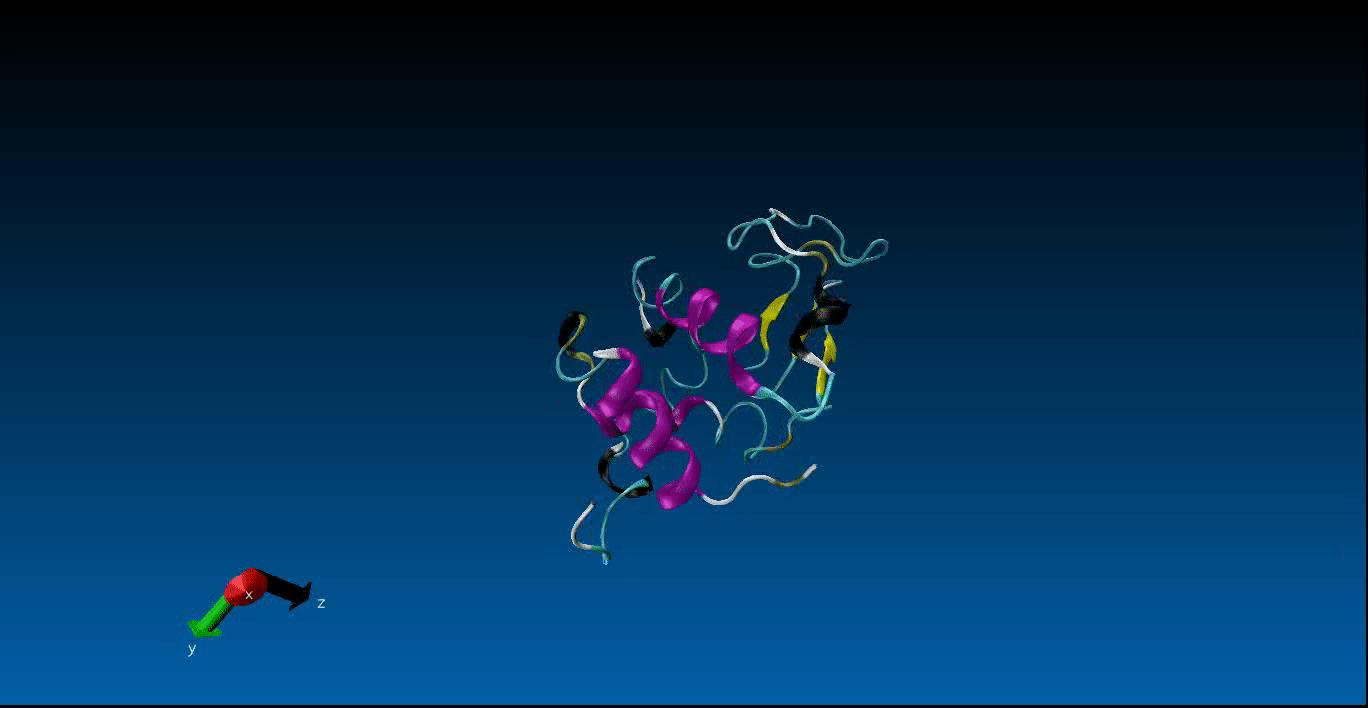
We are beginning our work in the area of protein design and the study of molecular dynamics. Currently we have configured the GROMACS package on our local machines together with pyRosetta. Below is a very short (10ns) simulation of the protein 1AKL using GROMACS that we have obtained. Stay tuned for more news in this area!
Medical Computing Research¶
We work on application of machine learning for identification of patterns in biomedical data (signals and images). Below is a list of some research projects in this area together with their summary and related publications.
Analysis of liver ultrasounds¶
Currently we are working on the analysis of liver texture and liver surface smoothness.
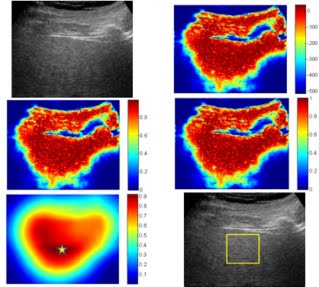
We have also developed a novel approach for detection of Fatty liver disease (FLD) and Heterogeneous liver using analysis of liver ultrasound images. The proposed system is able to automatically assign a representative region of interest (ROI) in a liver ultrasound which is subsequently used for diagnosis (as shown below). This ROI is analyzed using Wavelet Packet Transform (WPT) and a number of statistical features are obtained. A multi-class linear support vector machine (SVM) is then used for classification. The proposed system gives an overall accuracy of ~95%.
Automated Classification of Liver Disorders using Ultrasound Images, Fayyaz A. Afsar, Durr-e-Sabih, Mutawarra Hussain, in Journal of Medical Systems, vol. 34, no. 5, pp. 3163-3172, 2012 (Online), (Download draft)
Intelligent cardiac disease diagnosis¶
We have worked extensively on the development of ECG based detection of cardiac abnormalities as part of my MS (Systems Engineering) thesis and subsequent student projects. Below are the details of some this work. You can access more information about this work in my thesis.
ECG Acquisition¶
We have developed a hardware ECG acqusisition module which interfaces with PC or a Raspberry Pi through the USB. Below is a recording of the ECG we obtained using this hardware.
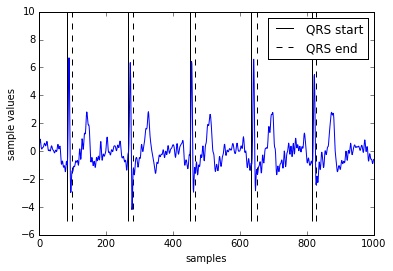
This hardware development was a part of the Abnormal Cardiac Beat Detection project.
ECG Preprocessing¶
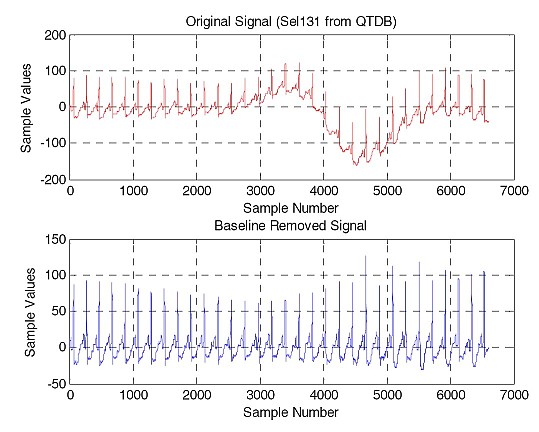
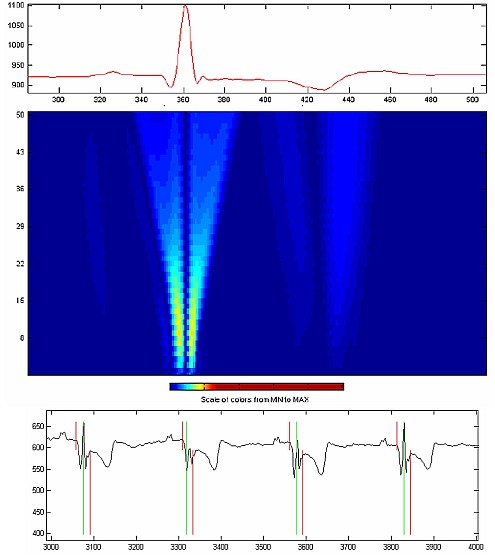
We have developed a number of techniques for noise and baseline removal from ECG signals. A number of wavelet based approaches were also implement for the delineation of ECG signals (breaking up the different parts of a single ECG beat).
- A Comparison of Baseline Removal Algorithms for Electrocardiogram (ECG) based Automated Diagnosis of Coronary Heart Disease, Fayyaz A. Afsar, M. S. Riaz and M. Arif, in proc. 3rd International Conference on Bioinformatics & Biomedical Engineering (iCBBE-2009), Beijing, PR of China, June 11-13, 2009. (Online)
- QRS Detection & Delineation for ECG Based Robust Clinical Decision Support System Design, Fayyaz A. Afsar, and M. Arif, in proc. National Research Conference 2007, Lahore, Pakistan, 2007 (Online).

Detection of Abnormal Beats¶
We have studied different types of features and classification schemes for the detection of abnormal beats in ECG.
- Pruned Fuzzy K-nearest neighbor Classifier for Beat Classification, M. Arif, M. Usman Akram, Fayyaz A. Afsar, in Journal of Biomedical Science and Engineering (JBISE), vol. 2010, no. 3, pp. 380-389, 2010. (Online)
- Robust Electrocardiogram (ECG) beat classification using Discrete Wavelet Transform, Fayyaz A. Afsar and M. Arif, in Physiological Measurement, 29 (2008), pp. 1-16, 2008. (Online)
Detection of Myocardial Infarction¶
We have used ensemble neural networks and principal components of ECG recordings to detection myocardial infarction in ECG data.
- Detection of ST Segment Deviation Episodes in the ECG using KLT with an Ensemble Neural Classifier, Fayyaz A. Afsar and M. Arif, in Physiological Measurement 29 (2008), pp. 747-760, 2008. (Online)
- Detection and Localization of Myocardial Infarction using K-Nearest Neighbor Classifier M. Arif, I.A. Malagore, Fayyaz .A. Afsar, Journal of Medical Systems, DOI 10.1007/s10916-010-9474-3, 2010. (Online)
Open Heart: ECG Based Cardiac Disease Analysis and Diagnosis System¶
You can read more about this projec below.
- Fayyaz A. Afsar, Open Heart: ECG Based Cardiac Disease Analysis and Diagnosis System, MS (System Engineering) thesis, Pakistan Institute of Engineering and Applied Sciences, Islamabad, Pakistan, 2007. (Download)
Machine Learning Research¶
Currently, we are working on multiple instance learning and feature selection.
We have developed pyLemmings: A software suite for Multiple Instance Learning and investigated Self-taught learning of the protein universe.
Below are some of our projects.
Biometrics¶
Fingerprint Identification¶
As part of my BS (Computer & Information Sciences) thesis, I developed a minutiae based fingerprint identification and classification system. We have developed our own fingerprints database.
- Fayyaz A. Afsar, Automatic Fingerprint Identification and Verification, BS (Computer and Information Sciences) thesis, Pakistan Institute of Engineering and Applied Sciences, Islamabad, Pakistan, 2005. (Download)
- Fingerprint Identification and Verification using Minutiae Matching, Fayyaz A. Afsar, M. Arif and M. Hussain, in proc. National Conference on Emerging Technologies, Karachi, Pakistan, 2004. (Download)
- Fingerprint Based Person Identification and Verification for Commercial Applications, Fayyaz A. Afsar, M. Arif, M. Hussain, in proc. IEEE International Multitopic Conference (INMIC) 2004, Lahore, Pakistan, 2004. (Online)
Speaker Recognition¶
I supervised an undergraduate student project aimed at telephonic, cellular phone and non-telephonic speaker recognition. We developed a speaker database for this purpose containing more than 60 speakers.
- Wavelet Transform Based Automatic Speaker Recognition, S. Malik and Fayyaz A. Afsar, in proc. International Multitopic Conference (INMIC), Islamabad, Pakistan, December 2010. (Online)
- Thesis by S. Malik: Download here.
Signature Recognition¶
I supervised an undergraduate student project aimed at person identification using online signatures. For this purpose, we developed a database of online signatures and their skilled forgeries for more 100 individuals.

- Thesis by M. Nauman Sajid: Download here.
- Wavelet Transform Based Global Features for Online Signature Recognition, Fayyaz A. Afsar, U. Farrukh and M. Arif, in proc. IEEE International Multitopic Conference (INMIC) 2005, Karachi, Pakistan, 2005 (Online).
Palm and Hand based Identification¶
As a student project I supervised, we developed a hand and palm based person identification system. We developed our own database of hand images for this purpose.
- Person Identification based on Palm & Hand Geometry, Qaiser N. Ashraf and Fayyaz A. Afsar, presented at Doctoral Symposium on Computer Science, Lahore, Pakistan, August 9-10, 2008. (Download)
- Thesis by Qaiser N. Ashraf: Download here.

Evolutionary algorithms¶
MOX¶
MOX is a novel computational intelligence approach to numerical optimization for finding the global optima of a multidimensional function based upon biological inspirations drawn from the highly selective behavior of female mosquitoes in choosing a habitat to lay their eggs and the inhibition of those eggs to hatch into the next stage. Comparison of convergence results of MOX for different benchmarks with existing methods in the literature clearly indicates the efficacy of the proposed scheme in maintaining a high success rate while reducing the number of function evaluations (NFE) required to reach a goal value for an optimization problem.
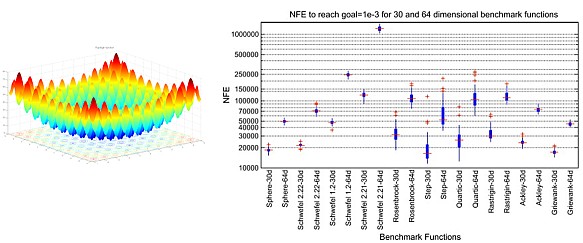
- MOX: A Novel Global Optimization Algorithm Inspired from Oviposition Site Selection and Egg Hatching Inhibition in Mosquitoes, Fayyaz A. Afsar and M. Arif in Journal of Applied Soft Computing, vol. 11, no. 8, pp. 4614-4625, 2011 (Paper), (Download draft), (Matlab Code)
GA based Robotic path planning¶
We developed a novel genetic algorithm based robotic path planning algorithm based on morphological processing.
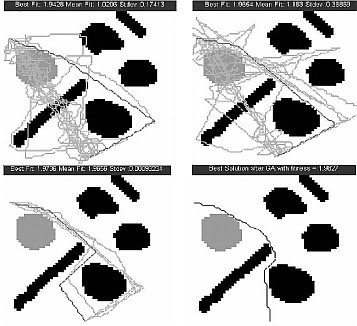
- Genetic Algorithm Based Path Planning and Optimization for Autonomous Mobile Robots with Morphological Preprocessing, Fayyaz A. Afsar, M. Arif, and M. Hussain, in proc. IEEE International Multitopic Conference (INMIC) 2006, Karachi, Pakistan, 2006. (Online)
Publications¶
You can visit our publications page here.
Software¶
You can view and use our software here.
Members¶
You can view details of current and past students here. We are also very thankful to all our collaborators.
The Logo¶
The logo of the lab shows the multidiciplinary nature of the research in the lab with biology, medicine and computing intertwined. This is depicted by the two wings: one made of feathers and the other using a binary tree which is widely used in computing. The DNA double helix shows the link of biology and computing with one strand made of nucleotides and the other showing a 4th order binary De-Bruijn sequence. The torch and the astrolobe on the top serve as symbols of scientific exploration. The urdu verse at the bottom is by The Poet of the East Dr. Muhammad Iqbal and it roughly translates to “Let nature face knowledge!”. The complete quatrain by Iqbal is given below.
فطرت کو خرد کے روبرو کر
تسخیر مقام رنگ بو کر
بے ذوق نہیں اگرچہ فطرت
جو اس سے نہ ہو سکا وہ تو کر
Let nature face knowledge!
Explore!
Nature is not untasteful by itself,
But you should do what it could not!